
Tutorials
The tutorial requires the mOTU profiler to be correctly installed as described in the Installation section.
General Workflow
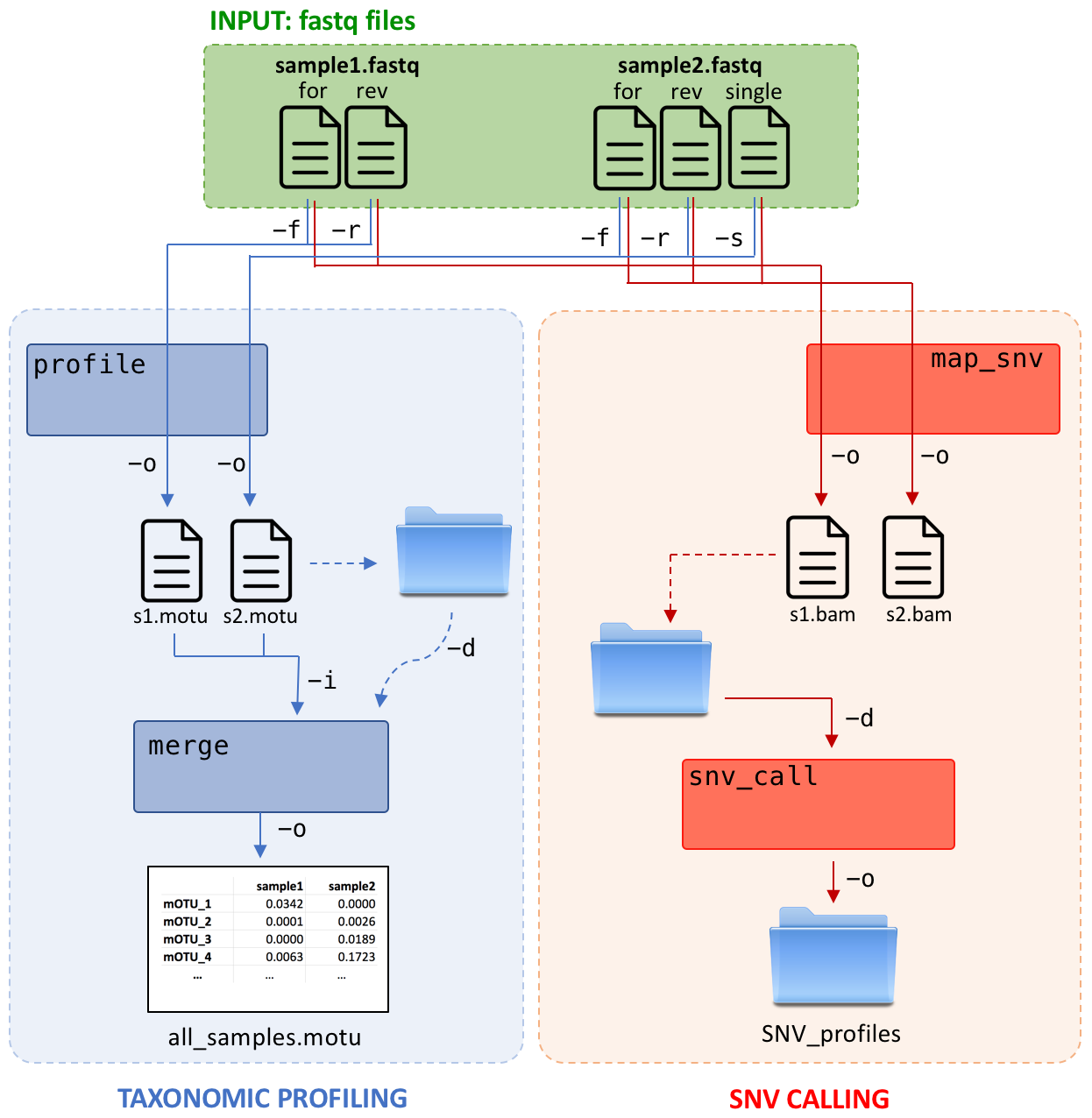
1. Generating taxonomic profiles
Standard example
Taxonomic profiles can be generated using the profile command and one or more sequencing files in fastq format:
# Using the mOTU profiler with test files contained in the installation/database directory
$motus profile -s mOTUs/db_mOTU/db_mOTU_test/test1_single.fastq -o test1.motus -n test1
$motus profile -s mOTUs/db_mOTU/db_mOTU_test/test1_single.fastq -o test1.motus -n test1
The resulting profile reports the relative abundance for each mOTU (ref + meta + ext):
$head -n 3 test1.motus
# git tag version 3.0.0 | motus version 3.0.0 | map_tax 3.0.0 | gene database: nr3.0.0 | calc_mgc 3.0.0 -y insert.scaled_counts -l 75 | calc_motu 3.0.0 -k mOTU -C no_CAMI -g 3 | taxonomy: ref_mOTU_3.0.0 meta_mOTU_3.0.0
# call: python mOTUs/motus profile -s mOTUs/db_mOTU/db_mOTU_test/test1_single.fastq
# consensus_taxonomy test1
$awk '{print $NF,$0}' test1.motus | sort -n -k 1 | cut -f 2- -d " " | tail -n 10
Pseudomonas sp. [ref_mOTU_v25_00201] 0.0237914022
Streptococcus sanguinis/cristatus [ref_mOTU_v25_01591] 0.0243451871
Francisella philomiragia/noatunensis [ref_mOTU_v25_02803] 0.0325841843
Corynebacterium ihumii/afermentans [ref_mOTU_v25_03430] 0.0429362887
Chlamydia abortus [ref_mOTU_v25_00757] 0.0584757145
Kandleria vitulina [ref_mOTU_v25_04327] 0.0755906022
Streptomyces durhamensis [ref_mOTU_v25_00648] 0.0796695140
Firmicutes species incertae sedis [meta_mOTU_v25_13597] 0.1116943104
Actinobacteria sp. [ref_mOTU_v25_03277] 0.1190951935
Streptococcus anginosus/intermedius [ref_mOTU_v25_00567] 0.3644241810
Read counting
The -c flag changes the output from relative abundance to number of assigned reads:
$motus profile -s mOTUs/db_mOTU/db_mOTU_test/test1_single.fastq -o test1.motus -n test1 -c
$head -n 3 test1.motus
# git tag version 3.0.0 | motus version 3.0.0 | map_tax 3.0.0 | gene database: nr3.0.0 | calc_mgc 3.0.0 -y insert.scaled_counts -l 75 | calc_motu 3.0.0 -k mOTU -C no_CAMI -g 3 -c | taxonomy: ref_mOTU_3.0.0 meta_mOTU_3.0.0
# call: python mOTUs/motus profile -s mOTUs/db_mOTU/db_mOTU_test/test1_single.fastq -c
#consensus_taxonomy test1
$awk '{print $NF,$0}' test1.motus | sort -n -k 1 | cut -f 2- -d " " | tail -n 10
Pseudomonas sp. [ref_mOTU_v25_00201] 11
Streptococcus sanguinis/cristatus [ref_mOTU_v25_01591] 12
Francisella philomiragia/noatunensis [ref_mOTU_v25_02803] 16
Corynebacterium ihumii/afermentans [ref_mOTU_v25_03430] 21
Chlamydia abortus [ref_mOTU_v25_00757] 28
Kandleria vitulina [ref_mOTU_v25_04327] 36
Streptomyces durhamensis [ref_mOTU_v25_00648] 38
Firmicutes species incertae sedis [meta_mOTU_v25_13597] 54
Actinobacteria sp. [ref_mOTU_v25_03277] 57
Streptococcus anginosus/intermedius [ref_mOTU_v25_00567] 175
Taxonomic level
The -k flag changes taxonomic level:
$motus profile -s mOTUs/db_mOTU/db_mOTU_test/test1_single.fastq -o test1.motus -n test1 -k phylum
$head -n 3 test1.motus
# git tag version 3.0.0 | motus version 3.0.0 | map_tax 3.0.0 | gene database: nr3.0.0 | calc_mgc 3.0.0 -y insert.scaled_counts -l 75 | calc_motu 3.0.0 -k phylum -C no_CAMI -g 3 | taxonomy: ref_mOTU_3.0.0 meta_mOTU_3.0.0
# call: python mOTUs/motus profile -s mOTUs/db_mOTU/db_mOTU_test/test1_single.fastq -k phylum
# consensus_taxonomy test1
$ awk '{print $NF,$0}' test1.motus | sort -n -k 1 | cut -f 2- -d " " | tail -n 10
candidate division Zixibacteria 0.0000000000
Thermodesulfobacteria 0.0061045472
unassigned 0.0101055004
Chlamydiae 0.0584757145
Proteobacteria 0.0775585823
Actinobacteria 0.2417009963
Firmicutes 0.6060546594
Threading
You can assign multiple cores (-t flag) to accelerate the alignment process:
$motus profile -f sample_R1.fq.gz -r sample_R2.fq.gz -t 8 -o test1.motus
Database selection
Add the -e flag to perform taxonomic profiling using only ref-mOTUs database:
$motus profile -f sample_R1.fq.gz -r sample_R2.fq.gz -e -o test1.motus
Merging profiles
mOTUs profiles from multiple samples can be merged into one combined profile. Also, this step can be used to add public profiles from 23 environments to your samples for comparison.
$motus merge -i test1.motus,test2.motus -o test.motus
$head -n 3 test.motus
# git tag version 3.0.0 | motus version 3.0.0 | map_tax 3.0.0 | gene database: nr3.0.0 | calc_mgc 3.0.0 -y insert.scaled_counts -l 75 | calc_motu 3.0.0 -k phylum -C no_CAMI -g 3 | taxonomy: ref_mOTU_3.0.0 meta_mOTU_3.0.0
# call: python mOTUs/motus profile -s mOTUs/db_mOTU/db_mOTU_test/test1_single.fastq -k phylum
# consensus_taxonomy test1 test2
$ awk '{print $NF,$0}' test.motus | sort -n -k 1 | cut -f 2- -d " " | tail -n 10
Pseudomonas sp. [ref_mOTU_v25_00201] 11 4
Streptococcus sanguinis/cristatus [ref_mOTU_v25_01591] 12 11
Francisella philomiragia/noatunensis [ref_mOTU_v25_02803] 16 30
Corynebacterium ihumii/afermentans [ref_mOTU_v25_03430] 21 19
Chlamydia abortus [ref_mOTU_v25_00757] 28 129
Kandleria vitulina [ref_mOTU_v25_04327] 36 1
Streptomyces durhamensis [ref_mOTU_v25_00648] 38 33
Firmicutes species incertae sedis [meta_mOTU_v25_13597] 54 12
Actinobacteria sp. [ref_mOTU_v25_03277] 57 13
Streptococcus anginosus/intermedius [ref_mOTU_v25_00567] 175 0
...
Merging your samples with public samples can be done by selecting 1 or many environments that should be added to your profiles. List of all environments:
- all
- air
- bioreactor
- bee
- cat
- cattle
- chicken
- dog
- fish
- freshwater
- human
- marine
- mouse
- pig
- sheep
- soil
- termite
- freshwater
# Adding bee samples to you profiles
$motus merge -i test1.motus,test2.motus -a bee -o test.motus
# Adding bee and dog samples to you profiles
$motus merge -i test1.motus,test2.motus -a bee,dog -o test.motus
# Adding all public profiles to your samples
$motus merge -i test1.motus,test2.motus -a all -o test.motus
More options
There are more options that influence the quality of the alignment (minimum length, percent identity) or change the output format and reported quantities (NCBI taxID, full rank, summarizing at a specific taxonomic rank). These options are displayed when
using the plain $motus profile command:
$motus profile
2. Generating metatranscriptomic profiles
Standard example
Taxonomic profiles can be generated using the profile command and one or more sequencing files in fastq format:
# Using the mOTU profiler with test files contained in the installation/database directory
$motus profile -s mOTUs/db_mOTU/db_mOTU_test/test1_single.fastq -o test1.motus -n test1
$motus profile -s mOTUs/db_mOTU/db_mOTU_test/test1_single.fastq -o test1.motus -n test1
The resulting profile reports the relative abundance for each mOTU (ref + meta + ext):
$head -n 3 test1.motus
# git tag version 3.0.0 | motus version 3.0.0 | map_tax 3.0.0 | gene database: nr3.0.0 | calc_mgc 3.0.0 -y insert.scaled_counts -l 75 | calc_motu 3.0.0 -k mOTU -C no_CAMI -g 3 | taxonomy: ref_mOTU_3.0.0 meta_mOTU_3.0.0
# call: python mOTUs/motus profile -s mOTUs/db_mOTU/db_mOTU_test/test1_single.fastq
# consensus_taxonomy test1
$awk '{print $NF,$0}' test1.motus | sort -n -k 1 | cut -f 2- -d " " | tail -n 10
Pseudomonas sp. [ref_mOTU_v25_00201] 0.0237914022
Streptococcus sanguinis/cristatus [ref_mOTU_v25_01591] 0.0243451871
Francisella philomiragia/noatunensis [ref_mOTU_v25_02803] 0.0325841843
Corynebacterium ihumii/afermentans [ref_mOTU_v25_03430] 0.0429362887
Chlamydia abortus [ref_mOTU_v25_00757] 0.0584757145
Kandleria vitulina [ref_mOTU_v25_04327] 0.0755906022
Streptomyces durhamensis [ref_mOTU_v25_00648] 0.0796695140
Firmicutes species incertae sedis [meta_mOTU_v25_13597] 0.1116943104
Actinobacteria sp. [ref_mOTU_v25_03277] 0.1190951935
Streptococcus anginosus/intermedius [ref_mOTU_v25_00567] 0.3644241810
Read counting
The -c flag changes the output from relative abundance to number of assigned reads:
$motus profile -s mOTUs/db_mOTU/db_mOTU_test/test1_single.fastq -o test1.motus -n test1 -c
$head -n 3 test1.motus
# git tag version 3.0.0 | motus version 3.0.0 | map_tax 3.0.0 | gene database: nr3.0.0 | calc_mgc 3.0.0 -y insert.scaled_counts -l 75 | calc_motu 3.0.0 -k mOTU -C no_CAMI -g 3 -c | taxonomy: ref_mOTU_3.0.0 meta_mOTU_3.0.0
# call: python mOTUs/motus profile -s mOTUs/db_mOTU/db_mOTU_test/test1_single.fastq -c
#consensus_taxonomy test1
$awk '{print $NF,$0}' test1.motus | sort -n -k 1 | cut -f 2- -d " " | tail -n 10
Pseudomonas sp. [ref_mOTU_v25_00201] 11
Streptococcus sanguinis/cristatus [ref_mOTU_v25_01591] 12
Francisella philomiragia/noatunensis [ref_mOTU_v25_02803] 16
Corynebacterium ihumii/afermentans [ref_mOTU_v25_03430] 21
Chlamydia abortus [ref_mOTU_v25_00757] 28
Kandleria vitulina [ref_mOTU_v25_04327] 36
Streptomyces durhamensis [ref_mOTU_v25_00648] 38
Firmicutes species incertae sedis [meta_mOTU_v25_13597] 54
Actinobacteria sp. [ref_mOTU_v25_03277] 57
Streptococcus anginosus/intermedius [ref_mOTU_v25_00567] 175
Taxonomic level
The -k flag changes taxonomic level:
$motus profile -s mOTUs/db_mOTU/db_mOTU_test/test1_single.fastq -o test1.motus -n test1 -k phylum
$head -n 3 test1.motus
# git tag version 3.0.0 | motus version 3.0.0 | map_tax 3.0.0 | gene database: nr3.0.0 | calc_mgc 3.0.0 -y insert.scaled_counts -l 75 | calc_motu 3.0.0 -k phylum -C no_CAMI -g 3 | taxonomy: ref_mOTU_3.0.0 meta_mOTU_3.0.0
# call: python mOTUs/motus profile -s mOTUs/db_mOTU/db_mOTU_test/test1_single.fastq -k phylum
# consensus_taxonomy test1
$ awk '{print $NF,$0}' test1.motus | sort -n -k 1 | cut -f 2- -d " " | tail -n 10
candidate division Zixibacteria 0.0000000000
Thermodesulfobacteria 0.0061045472
unassigned 0.0101055004
Chlamydiae 0.0584757145
Proteobacteria 0.0775585823
Actinobacteria 0.2417009963
Firmicutes 0.6060546594
Threading
You can assign multiple cores (-t flag) to accelerate the alignment process:
$motus profile -f sample_R1.fq.gz -r sample_R2.fq.gz -t 8 -o test1.motus
Database selection
Add the -e flag to perform taxonomic profiling using only ref-mOTUs database:
$motus profile -f sample_R1.fq.gz -r sample_R2.fq.gz -e -o test1.motus
Merging profiles
mOTUs profiles from multiple samples can be merged into one combined profile. Also, this step can be used to add public profiles from 23 environments to your samples for comparison.
$motus merge -i test1.motus,test2.motus -o test.motus
$head -n 3 test.motus
# git tag version 3.0.0 | motus version 3.0.0 | map_tax 3.0.0 | gene database: nr3.0.0 | calc_mgc 3.0.0 -y insert.scaled_counts -l 75 | calc_motu 3.0.0 -k phylum -C no_CAMI -g 3 | taxonomy: ref_mOTU_3.0.0 meta_mOTU_3.0.0
# call: python mOTUs/motus profile -s mOTUs/db_mOTU/db_mOTU_test/test1_single.fastq -k phylum
# consensus_taxonomy test1 test2
$ awk '{print $NF,$0}' test.motus | sort -n -k 1 | cut -f 2- -d " " | tail -n 10
Pseudomonas sp. [ref_mOTU_v25_00201] 11 4
Streptococcus sanguinis/cristatus [ref_mOTU_v25_01591] 12 11
Francisella philomiragia/noatunensis [ref_mOTU_v25_02803] 16 30
Corynebacterium ihumii/afermentans [ref_mOTU_v25_03430] 21 19
Chlamydia abortus [ref_mOTU_v25_00757] 28 129
Kandleria vitulina [ref_mOTU_v25_04327] 36 1
Streptomyces durhamensis [ref_mOTU_v25_00648] 38 33
Firmicutes species incertae sedis [meta_mOTU_v25_13597] 54 12
Actinobacteria sp. [ref_mOTU_v25_03277] 57 13
Streptococcus anginosus/intermedius [ref_mOTU_v25_00567] 175 0
...
Merging your samples with public samples can be done by selecting 1 or many environments that should be added to your profiles. List of all environments:
- all
- air
- bioreactor
- bee
- cat
- cattle
- chicken
- dog
- fish
- freshwater
- human
- marine
- mouse
- pig
- sheep
- soil
- termite
- freshwater
# Adding bee samples to you profiles
$motus merge -i test1.motus,test2.motus -a bee -o test.motus
# Adding bee and dog samples to you profiles
$motus merge -i test1.motus,test2.motus -a bee,dog -o test.motus
# Adding all public profiles to your samples
$motus merge -i test1.motus,test2.motus -a all -o test.motus
More options
There are more options that influence the quality of the alignment (minimum length, percent identity) or change the output format and reported quantities (NCBI taxID, full rank, summarizing at a specific taxonomic rank). These options are displayed when
using the plain $motus profile command:
$motus profile
3. Generating single nucleotide variant (SNV) profiles using MGs
Calling variants using marker genes is divided in to two subroutines, namely alignment and variant calling (map_snv, snv_call). map_snv aligns sequencing reads against the mOTU profiler database. snv_call utilizes the metaSNV package to call variants on these marker genes.
map_snv takes one or multiple sequencing files and aligns reads against the mOTU profiler database:
motus map_snv -s sample.fq.gz > sample.bam
motus map_snv -f sample_R1.fq.gz -r sample_R2.fq.gz > sample.bam
motus map_snv -f sample_R1.fq.gz -r sample_R2.fq.gz > sample.bam
Tweaking alignment parameters allows for changes in the minimum alignment length (-l). The -t flag allows to accelerate the alignment step using multithreading:
motus map_snv -f sample_R1.fq.gz -r sample_R2.fq.gz -l 100 -t 8> sample.bam
snv_call takes the bam files created in the map_snv step as input and calls variants using the metaSNV package. This information is then be used to create a distance matrix between samples. The input for snv_call is a directory with bam files. Each bam
file will be treated as an individual sample:
motus snv_call -d DIRECTORY -o OUTPUT_DIRECTORY
An example distance matrix for the comparison of 3 samples is shown below.
-------- sample_1 sample_2 sample_3
sample_1 0.0000 0.0012 0.1430
sample_2 0.0012 0.0000 0.1392
sample_3 0.1430 0.1392 0.0000
sample_1 0.0000 0.0012 0.1430
sample_2 0.0012 0.0000 0.1392
sample_3 0.1430 0.1392 0.0000
There are multiple filtering parameters that influence if variants are called such as coverage depth (-fd), coverage breadth (-fb) or the minimum number of samples that report a variant (-fm). A list of all parameter can be found when executing the plain
motus snv_call command:
motus snv_call
