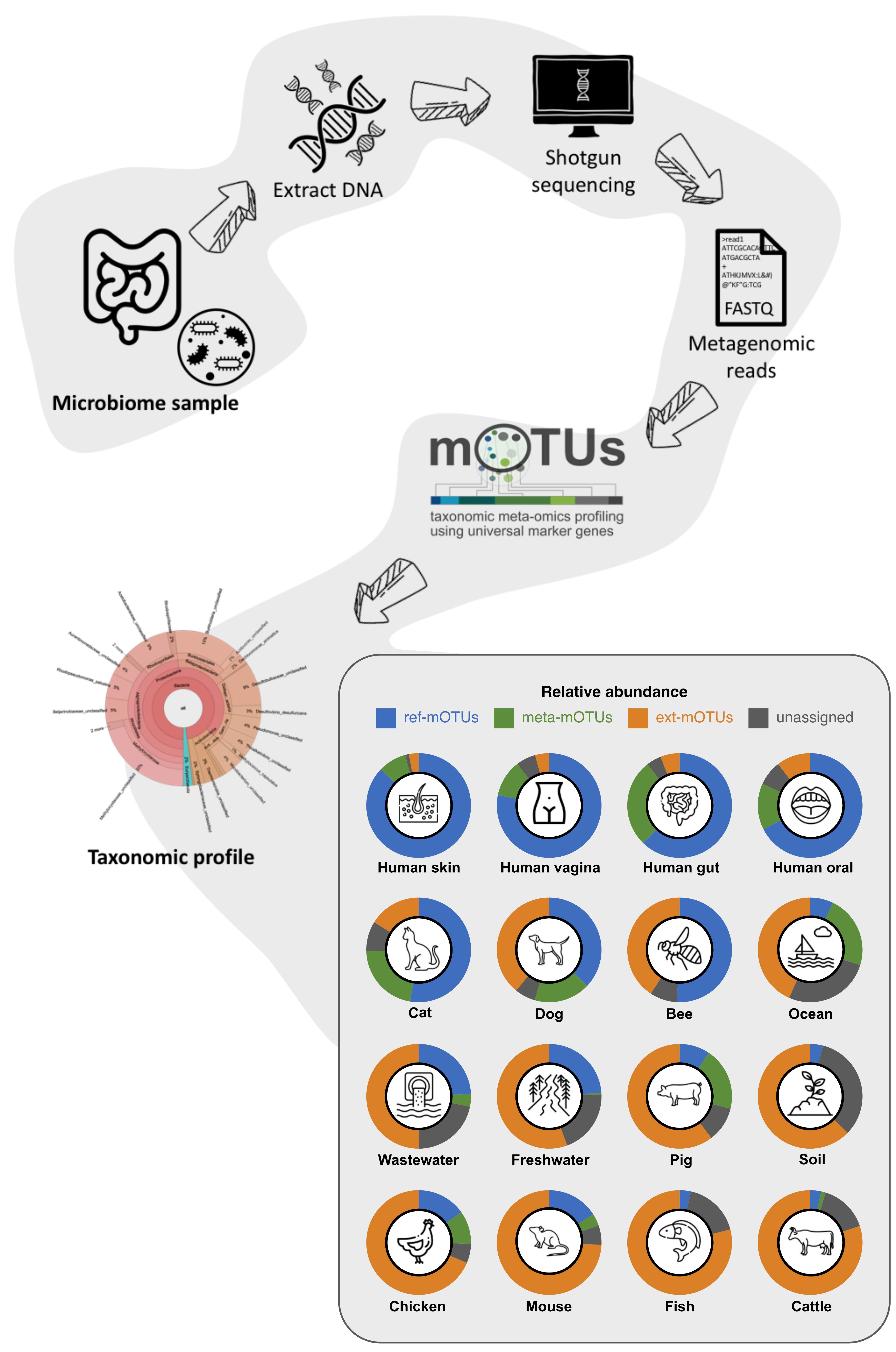
To profile a metagenomic or metatranscriptomic sample, install the tool and follow the instructions in the tutorial. If you use this tool in published work, please cite us.
Summary
Phylogenetic markers are genes that can be used to reconstruct the evolutionary history of organisms and to profile the taxonomic composition of environmental samples. Efforts to find a good set of protein-coding phylogenetic marker genes led to the identification of 40 universal marker genes (MGs) [1,2]. These 40 MGs occur in single copy in the vast majority of known organisms and they have been used to delineate prokaryotic organisms at the species level [3]. The MG-based operational taxonomic unit (mOTU) profiler, originally described in [4], uses 10 of the 40 MGs to taxonomically profile shotgun metagenomes, to quantify metabolically active members in metatranscriptomics and to quantify differences between strain populations using single nucleotide variation (SNV) profiles [5]. To generate the mOTUs database, we extracted MGs from ~87,000 prokaryotic reference genomes and over 3,100 publicly available metagenomes from major human body sites, gut microbiome samples from disease association studies, and ocean water samples. The clustering of MGs resulted in 11,915 reference mOTUs (ref-mOTUs) and 2,297 metagenomic mOTUs (meta-mOTUs) [5]. For mOTUs 3, we extended the database by 19,358 ext-mOTUs that originated from ~600,000 metagenome-assembled genomes spanning 23 environments, such as mouse, cat, dog, pig, freshwater, wastewater, air, and many others [6].